Genome annotation
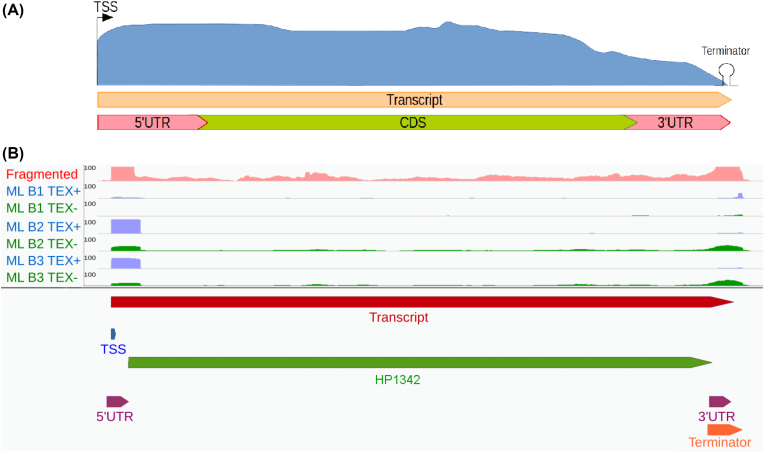
In order to interpret and translate omics datasets into biological and medical insights. Genome annotation is a basic requirement. We implemented ANNOgesic - the first tool for bacterial/archaeal RNA-seq-based genome annotations (S.H. Yu, 2018, GigaScience). ANNOgesic was reported and collected on many platforms and websites. Currently, we are continuing to extend ANNOgesic to more applications and make it more user-friendly for scientists who have no bioinformatic background. Moreover, an annotation tool for eukaryotic species is also being developed.
Multi-Omics data analysis
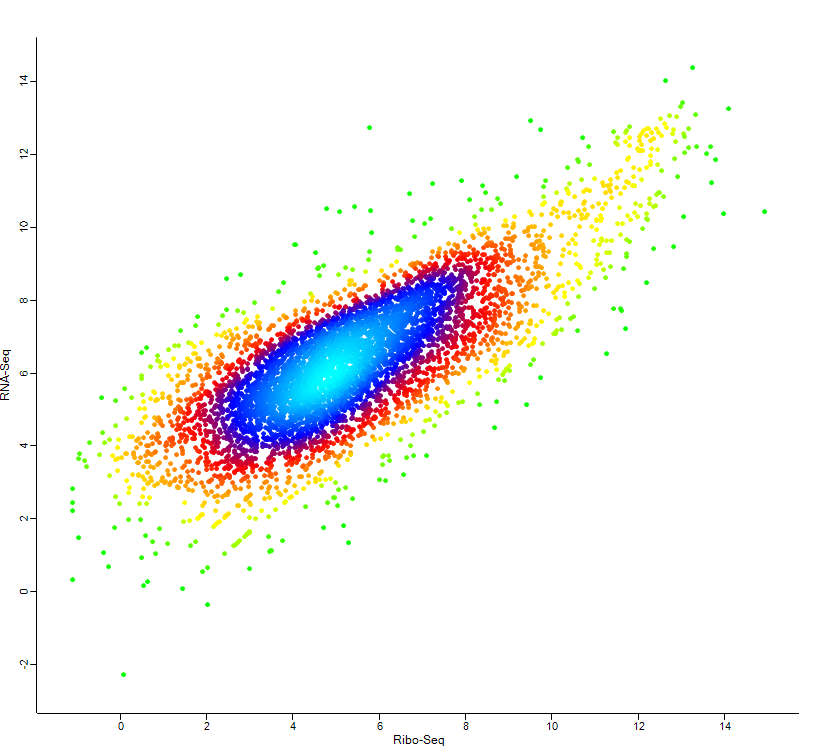
High-throughput technology revolutionized the whole field of biology. Numerous analyzing tools for NGS and proteomics data have been published in recent years. However, a single omics data may not reveal a complete picture for the global understanding. Every omics has different advantages but contains some blind spots as well. For instance, transcriptomics can easily detect non-coding genes but is not a proper candidate for the studies of phenotypes. Integration of multi-omics data will definitely be the discernible trend in the following years. Although the global correlation between transcriptomics and proteomics data is quite low (~10-25%), many studies still successfully used multi-omics to achieve specific goals like protein/RNA complex. Thus, the aims of our research in multi-omics analysis go in two directions – improvement of the global integration for multi-omics data and discovery of the issues that can be solved by multi-omics.
Single cell transcriptomic and proteomics

Due to the rapid progress of next-generation sequencing technologies, studying the characterization of individual cells can be achieved. Single-cell technology is the only way to understand biological heterogeneity or study rare cells. In recent years, the number of single-cell studies in genome and transcriptome levels has increased significantly, and the technology is reaching maturity as well. However, single-cell proteomics is still in the initial stage, and numerous possible projects can still be explored. we are involved in some projects with outstanding scientists in the single-cell proteomics and transcriptomics community (S.H. Yu., 2020, J. Proteome Res.). The aim of the projects is to develop a useful tool for single-cell proteomics and transcriptomics.
Epigenomics and DNA methylation
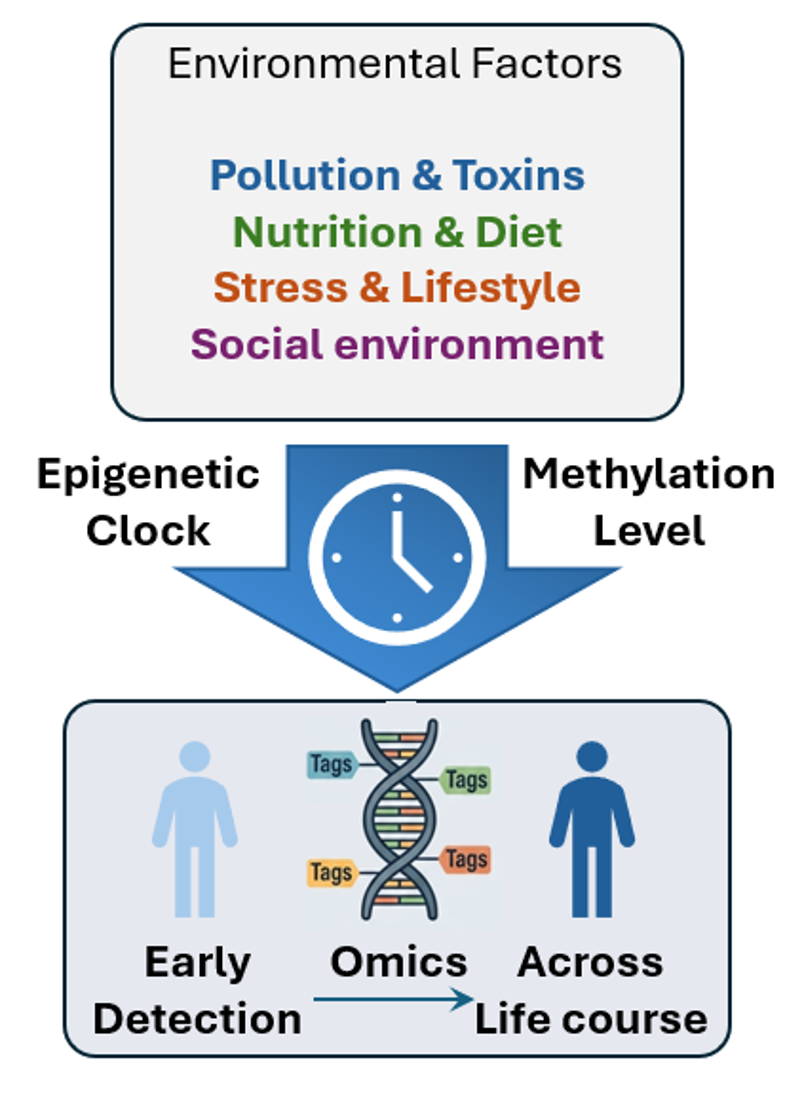
Epigenomics studies the dynamic chemical modifications on our DNA—such as DNA methylation—that turn genes "on" or "off" without altering the underlying genetic code. Acting as a vital bridge between environment and biology, these molecular tags adapt to factors like diet, stress, and toxins, leaving lasting marks that often accumulate from early development. Because these changes track predictably over time, scientists use them to build epigenetic clocks that measure biological aging and chronic disease risk. Ultimately, mapping these methylation patterns serves as a powerful tool for early disease detection, allowing clinicians to intercept conditions like cancer long before physical symptoms appear. To translate these insights into clinical tools, our team is actively developing and applying novel epigenetic clock methodologies to measure biological aging and predict chronic disease risk. Ultimately, by mapping these precise environmental and genomic influences, our work aims to pioneer breakthrough strategies for early disease detection and precision medicine.
Deep learning/Machine learning
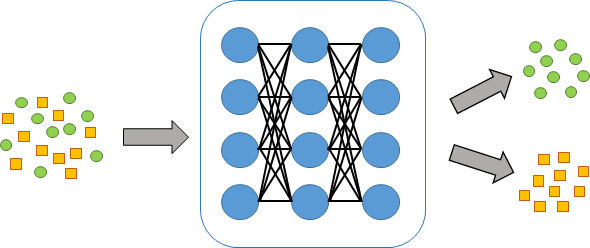
Deep learning is a subset of artificial intelligence which have caught a lot of attention recently. Currently, many studies show that deep learning performs high-quality and accurate predictions in the bioinformatics field. However, applying deep learning technology to omics data analysis is still fresh ground that needs to be reclaimed. The goal of the project is to build a precise tool for analyzing omics data.